Submitting a new structure
We encourage the active submission of newly determined amyloid fibril structures. Users can submit a new structure by using the database submission form. To access this form, click on the submit button which can be found on the top-right corner next to the database menu button. Users are free to submit new files in PDF format, along with information on the protein name and PDB ID (or as a zip file). New submissions will then be manually curated and automatically analysed before being released as new entries.
Protein entries
Sequence section
- This section in protein entries highlights the WT protein sequence, with individual residues shown as boxes
- Residues corresponding to experimentally determined aggregation prone regions are shown as shaded boxes
- The sequence lengths of selected amyloid structural polymorphs are dynamically noted with blue and purple coloured linings at the top and bottom of corresponding residue boxes
- Disease-related mutations are colour-coded based on polymorph selection and shown as extra letters in the corresponding WT residue box positions
3D models
Side-by-side panels of molecular graphic interfaces can be used to perform pairwise structural comparisons of selected polymorphs. Amyloid polymorphs are automatically loaded after user selection in each graphical interface, which support multiple visualisation options for graphical representation of the structural models (more than 21 representation modes available) and for property-based colour-coding schemes.
Thermodynamic profiling
We utilise the FoldX energy force field to calculate the energy contribution of each individual residue on the stability of the amyloid fold for each polymorph structure (Van der Kant R et al. bioRxiv 2021.03.01.433317).
FoldX first calculates the free energy contribution of each atom in the protein based on its position relative to its neighbours in the structure. These contributions are then summed, first at the residue level and later to the level of the entire protein. This allows to map the contribution of each residue to the total free energy of the protein (called ΔGcontrib), but also to report on individual thermodynamic components, such as the sum of contributions from Van der Waals interactions (ΔGvdw), solvation energies for polar and apolar groups (ΔGsolvP and ΔGsolvH), electrostatics (ΔGel), H-bonding (ΔGHbond) and entropic penalties for main and side chains (ΔSmc and ΔSsc), as well as water H-bonding (ΔGwb). Overall contributions to structural stability were calculated based on the following function:
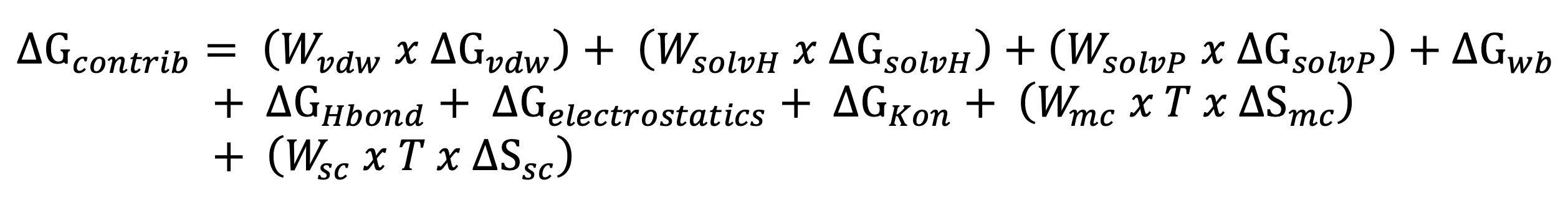
where the corresponding weights are set to 1, with the exception of the Wvdw which is set to 0.33, respectively.
The accuracy of the free energy estimation by FoldX correlates with structure quality parameters, such as crystallographic resolution. The quality of protein polypeptide backbone conformations can easily be estimated by plotting the Ramachandran map of these structures and evaluating whether backbone traces deviate from the allowed conformational space of each of its residues. In order to do this in a quantitative manner, FoldX uses a statistical thermodynamics approach based on the observed frequency distribution of each amino acid in the Ramachandran map in crystal structures of the highest resolution (selected using the WHATIF tool).
Residues with ΔGcontrib values (shown in the y-axis) below 0 are considered to contribute to the stability of the amyloid fold, whereas residues with ΔGcontrib values above 0 are considered as structural frustrated regions of the amyloid core.
__